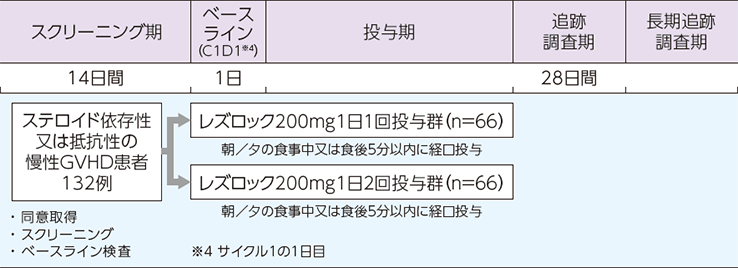
試験概要
†本試験において事前規定された解析時点(治験総括報告書におけるデータカットオフ日)は最終患者登録後6ヵ月経過時点ですが、本情報は承認審査過程で評価された解析時点(データカットオフ日)である最終患者登録後1年経過時点のデータに基づき作成しています。
(1)最良全奏効率(best ORR)(最終患者登録後1年経過時点†)
(主要評価項目):検証的な解析結果
best ORR(最終患者登録後1年経過時点)は、1日1回投与群、1日2回投与群でそれぞれ72.7%(48/66例)、77.3%(51/66例)であった。その95%CIはそれぞれ[60.4, 83.0]、[65.3, 86.7]であり、95%CIの下限値が事前に設定した有効性の判断基準(閾値30%)を上回った。なお、CRは1日1回投与群、1日2回投与群でそれぞれ6.1%(4/66例)、4.5%(3/66例)、PRはそれぞれ66.7%(44/66例)、72.7%(48/66例)であった。
best ORR(最終患者登録後1年経過時点)(mITT集団)
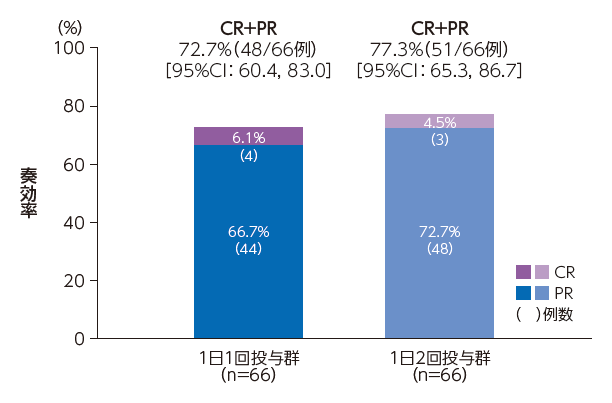
判断基準:best ORRの両側95%CI(Clopper Pearson[正確]法)の下限値が閾値30%を上回った場合
に有効と判断する。
最良総合効果
評価判定別割合(n)
LR:無効、LR-U:無効(不変)、LR-M:無効(改善と悪化の混合)、LR-P:無効(悪化)
†本試験において事前規定された解析時点(治験総括報告書におけるデータカットオフ日)は最終患者登録後6ヵ月経過時点ですが、本情報は承認審査過程で評価された解析時点(データカットオフ日)である最終患者登録後1年経過時点のデータに基づき作成しています。
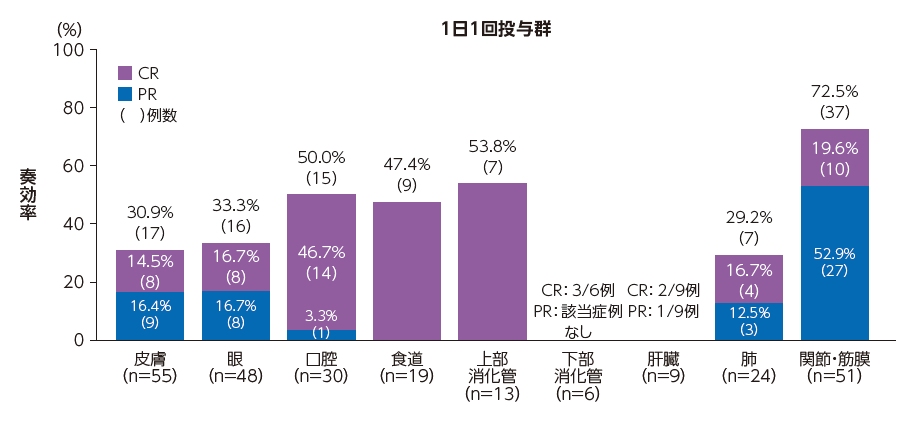
(2)臓器別奏効率(副次評価項目)
評価対象とした罹患臓器のいずれにおいても奏効(PR以上)が認められた。
臓器別奏効率※(mITT集団)
※下部消化管(1日1回投与群:n=6、1日2回投与群:n=7)、肝臓(1日1回投与群:n=9、1日2回投与群:n=4)は10例未満のため、%表記及びグラフ化は行っていない。
(3)安全性
安全性の結果概要(安全性解析対象集団)
1日1回投与群
65
(98.5) |
49
(74.2) |
主な副作用は疲労17例(25.8%)、悪心9例(13.6%)、下痢、嘔吐、頭痛が各6例(9.1%)であった。 |
27
(40.9) |
5
(7.6) |
重篤な副作用は5例(蜂巣炎、感染性大腸炎、ブドウ球菌性菌血症、肺炎が各1例、下痢、悪心、嘔吐及び多臓器機能不全症候群※1を併発した1例)に認められた。
※1 死亡症例と同一症例 |
16
(24.2) |
9
(13.6) |
投与中止に至った副作用は9例(悪心3例、疲労、蜂巣炎、好中球減少症、筋痙縮、開口障害※2、食欲減退※3、多臓器機能不全症候群※1、トランスアミナーゼ上昇が各1例)に認められた。
※1 死亡症例と同一症例
※2 筋痙縮を発現した症例と同一症例
※3 悪心を発現した症例と同一症例 |
4
(6.1) |
1
(1.5) |
死亡に至った副作用は1例(多臓器機能不全症候群)に認められた。 |
n(%)
1日2回投与群
66
(100.0) |
40
(60.6) |
主な副作用は疲労14例(21.2%)、悪心7例(10.6%)、AST増加6例(9.1%)であった。 |
23
(34.8) |
2
(3.0) |
重篤な副作用は2例(慢性移植片対宿主病※1、微小血管症性溶血性貧血が各1例)に認められた。
※1 死亡症例と同一症例 |
12
(18.2) |
7
(10.6) |
投与中止に至った副作用は7例(AST増加、頭痛、潰瘍性角膜炎、微小血管症性溶血性貧血、疲労、激越が各1例、ALT増加及びAST増加を併発した1例)に認められた。 |
4
(6.1) |
1
(1.5) |
死亡に至った副作用は1例(慢性移植片対宿主病)に認められた。 |
n(%)
AST:アスパラギン酸アミノトランスフェラーゼ、ALT:アラニンアミノトランスフェラーゼ
主な有害事象及び副作用の内訳(いずれかの群で有害事象発現率10%以上)
1日1回投与群及び1日2回投与群
MedDRA/J version 20.0
n(%)
Grade:NCI-CTCAE v5.0
AST:アスパラギン酸アミノトランスフェラーゼ、ALT:アラニンアミノトランスフェラーゼ
器官別大分類(SOC)では、基本語(PT)を複数事象発現した症例は1例としてカウントした。
同一事象が同一患者に複数回発現した場合は、Gradeが最も高い事象を1回のみカウントした。
特に関心のある安全性評価項目
1日1回投与群
n(%)
※ 治験薬との関連性ありと判定された有害事象は認められなかった。
1日2回投与群
n(%)
※ 治験薬との関連性ありと判定された有害事象は認められなかった。
AST:アスパラギン酸アミノトランスフェラーゼ、ALT:アラニンアミノトランスフェラーゼ
- 6. 用法・用量
通常、成人及び12歳以上の小児にはベルモスジルとして200mgを1日1回食後に経口投与する。併用薬に応じて、効果不十分な場合に1回200mg1日2回投与に増量できる。
- 7. 用法・用量に関連する注意
- 7.1 食後投与に比べて空腹時投与で本剤のCmax及びAUCが低下するため、本剤は食後に服用すること。[16.2.1 参照]
- 7.2 プロトンポンプ阻害剤又は強いCYP3A4誘導剤との併用により、本剤の血中濃度が低下する可能性があるため、これらの薬剤を併用する場合は患者の状態に注意し、本剤の効果が不十分な場合には、本剤を1回200mg1日2回投与に増量することを考慮すること。[10.2、16.7.1-16.7.3 参照]
- 8. 重要な基本的注意(一部抜粋)
- 8.2 肝機能障害があらわれることがあるので、本剤投与中は定期的に肝機能検査を実施すること。
- 9. 特定の背景を有する患者に関する注意(一部抜粋)
- 9.3 肝機能障害患者
- 9.3.1 重度(Child-Pugh分類C)の肝機能障害患者
可能な限り投与を避けること。やむを得ず投与する場合には、患者の状態をより慎重に観察し、有害事象の発現に十分注意すること。本剤の血中濃度が上昇し、副作用が強くあらわれるおそれがある。[16.6.1 参照]
2)社内資料:ベルモスジルの国内第Ⅲ相試験(ME3208-2試験)(承認時評価資料)
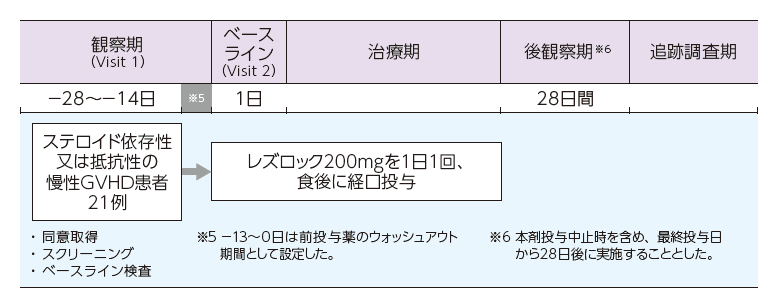
試験概要
本情報は、データカットオフ日(最終患者登録後24週経過時)時点のデータに基づき作成した。
allo-HCT:allogeneic hematopoietic cell transplantation、
%FEV1:percent predicted forced expiratory volume in one second、
CTCAE:Common Terminology Criteria for Adverse Events、
JCOG:Japan Clinical Oncology Group、
SMQ:standardised MedDRA queries(MedDRA 標準検索式)、
HLGT:high level group terms(高位グループ語)、
HLT:high level terms(高位語)
(1)最良全奏効率(best ORR)(最終患者登録後24週経過時点)
(主要評価項目):検証的な解析結果
best ORR(最終患者登録後24週経過時点)は85.7%(18/21例)であった。その95%CIは[63.66,96.95]であり、95%CIの下限値が事前に設定した有効性の判断基準(閾値25%)を上回った。なお、奏効が得られた患者の最良全奏効は、すべてPRであった。
best ORR(最終患者登録後24週経過時点)(mITT集団、PPS集団)
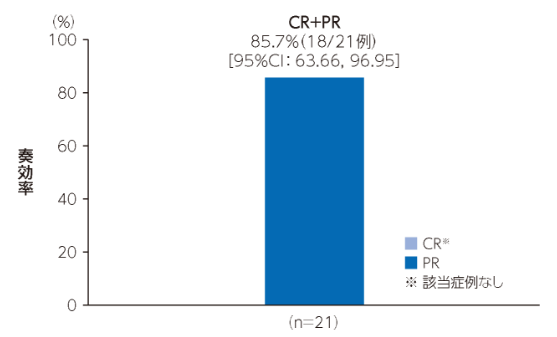
判断基準:best ORRの両側95%CI(Clopper Pearson[正確]法)の下限値が閾値25%を上回
った場合に有効と判断する。
最良総合効果
評価判定別割合(n)
LR:無効、LR-U:無効(不変)、LR-M:無効(改善と悪化の混合)、LR-P:無効(悪化)
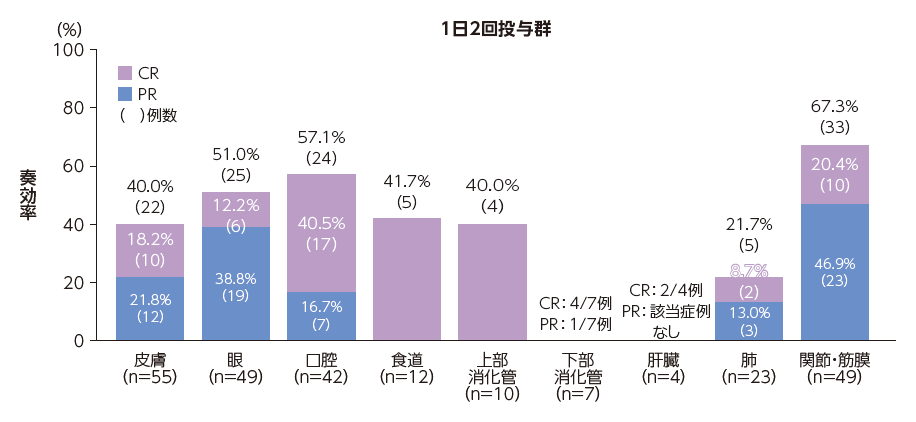
(2)臓器別奏効率(副次評価項目)
臓器別奏効率は、皮膚54.5%(6/11例)、眼20.0%(3/15例)、口腔66.7%(12/18例)、食道が2例中1例、上部消化管が1例中1例、関節・筋膜が5例中4例であった。下部消化管及び肺では奏効が認められなかった。なお、本試験に登録された患者において肝臓に病変を有する患者は含まれなかった。
臓器別奏効率(mITT集団、PPS集団)
最良総合効果
n(%)
※ 本試験に登録された患者において肝臓に病変を有する患者は含まれなかった。
(3)安全性(安全性解析対象集団)
安全性の結果概要
有害事象及び副作用の内訳
MedDRA/J version 24.1
n(%)
Grade:CTCAE v5.0-JCOG
器官別大分類(SOC)では、基本語(PT)を複数事象発現した症例は1例としてカウントした。
同一事象が同一患者に複数回発現した場合は、Gradeが最も高い事象を1回のみカウントした。
特に関心のある安全性評価項目
n(%)
※ 治験薬との関連性ありと判定された有害事象は認められなかった。
- 4. 効能・効果
造血幹細胞移植後の慢性移植片対宿主病(ステロイド剤の投与で効果不十分な場合)
- 6. 用法・用量
通常、成人及び12歳以上の小児にはベルモスジルとして200mgを1日1回食後に経口投与する。併用薬に応じて、効果不十分な場合に1回200mg1日2回投与に増量できる。
- 7. 用法・用量に関連する注意
- 7.1 食後投与に比べて空腹時投与で本剤のCmax及びAUCが低下するため、本剤は食後に服用すること。[16.2.1 参照]
- 7.2 プロトンポンプ阻害剤又は強いCYP3A4誘導剤との併用により、本剤の血中濃度が低下する可能性があるため、これらの薬剤を併用する場合は患者の状態に注意し、本剤の効果が不十分な場合には、本剤を1回200mg1日2回投与に増量することを考慮すること。[10.2、16.7.1-16.7.3 参照]
- 8. 重要な基本的注意(一部抜粋)
- 8.2 肝機能障害があらわれることがあるので、本剤投与中は定期的に肝機能検査を実施すること。
- 9. 特定の背景を有する患者に関する注意(一部抜粋)
- 9.3 肝機能障害患者
- 9.3.1 重度(Child-Pugh分類C)の肝機能障害患者
可能な限り投与を避けること。やむを得ず投与する場合には、患者の状態をより慎重に観察し、有害事象の発現に十分注意すること。本剤の血中濃度が上昇し、副作用が強くあらわれるおそれがある。[16.6.1 参照]
 ・
会員規約、および、
medパス利用規約・プライバシーポリシー
・
会員規約、および、
medパス利用規約・プライバシーポリシー の同意をお願いしております。
の同意をお願いしております。